
【数据竞赛】天池蛋白质结构预测大赛总结
共 5377字,需浏览 11分钟
·
2021-04-08 11:13
比赛名称:蛋白质结构预测大赛
比赛链接:https://tianchi.aliyun.com/competition/entrance/231781/information
比赛类型:自然语言处理
分享内容:Top5分享
比赛背景
蛋白质是生命活动中重要的组成,蛋白质的结构决定了蛋白质在生命活动中的功能,因此对蛋白质结构进行分析具有重要的实际意义。蛋白质结构又可分为一级结构,二级结构,三级结构和四级结构。
目前已知一部分氨基酸序列和与其对应的二级结构,通过已有数据寻找一级结构到二级结构的映射模型,提高通过氨基酸序列进行蛋白质二级结构预测的准确性。
比赛数据
对应的蛋白质氨基酸序列与二级结构序列共20000组,氨基酸序列和二级结构序列分别按行对应存放于data_seq_train和data_sec_train文件中构成训练数据,蛋白质序列长度不尽相同,每条蛋白质序列与二级结构序列长度相同,蛋白质序列中字母表示氨基酸类型,二级结构字母对应当前氨基酸构成的二级结构,二级结构中的空白也是一种松散结构的表示。
数据样本:
氨基酸序列:GIVEQCCTSICSLYQLENYCN 二级结构序列:TTTTSSS HHHHHTTB
Top1方案分享
分享原文:https://tianchi.aliyun.com/forum/postDetail?postId=98363
开源代码:https://github.com/wudejian789/2020TIANCHI-ProteinSecondaryStructurePrediction-TOP1
赛题理解
本题为根据蛋白质的一级结构预测其二级结构,经过比赛期间组内师兄的讲解,我对蛋白质一级结构二级结构的理解如下,如有错误,欢迎指正。
蛋白质可以看成是一条氨基酸序列,在空间中是一种相互交错螺旋的结构,像一条互相缠绕的绳子:
这种三维结构叫做蛋白质的三级结构,而如果不考虑结构的三维性,或者说把这整条序列拉直,用一个一维的序列表示,这便是得到了蛋白质的一级结构:
GPTGTGESKCPLMVKVLDAV······
这些字母G、A、V等便是代表一个个的氨基酸,其中主要包含有20种常见的氨基酸。
用这样的序列表示蛋白质比起原始的三维结构确实方便不少,但却丢失了三维的结构信息,蛋白质的结构决定其功能,这里的结构不止是序列本身,更多的还依赖其三维结构。
因此便出现了蛋白质的二级结构,它是一条与一级结构长度相等的一维序列,用以表征一级结构种的各位置的氨基酸在三维空间种的形态,以保留一部分的三维结构信息。
这里的' '、'E'、'T'等都是对应位置的氨基酸在空间种的形态(与一级结构 GPTGTGESKCPLMVKVLDAV······ 是一一对应的),例如'T'代表的就是该位置的氨基酸在空间中是一种氢键转折的形态。
本赛题就是需要通过蛋白质的一级结构,预测其二级结构,在深度学习种是一种典型的N-N的seq2seq问题。
赛题理解
不难想到,蛋白质三维结构的形成,其实主要是受某些力的作用,不同氨基酸的分子量、体积、质量等性质都有差异,这些小分子间会受到分子间作用力的影响,换句话说,分子间作用力等多种因素共同作用,让蛋白质形成了这样的一种相对稳定的空间结构,以达到一种稳态;而倘若你强行把它拉直,它也会由于受力不均,又开始相互缠绕,以达到稳态。
因此,对于某条蛋白质的二级结构中第i个位置的空间形态,其不止是取决于对应一级结构中位置i的氨基酸,还取决于位置i周围氨基酸甚至整条序列的情况。
定义一级结构中位置i及其上下文的整条片段为X,对应的二级结构中位置i的形态为Y,我统计了整个训练数据中 P(Y|X) 的情况,并计算了在不同窗口大小时。
思路分享
这类题首先需要解决的是输入序列的编码问题,很自然的可以想到onehot和word2vec两种编码方法,本次赛题我们都进行了尝试。
Onehot与基本理化性质编码+滑窗法+浅层NN
氨基酸的基本理化性质包括分子量、等电点、解离常数、范德华半径、水中溶解度、侧脸疏水性,以及形成α螺旋可能性、形成β螺旋可能性、转向概率等(来自Chou-Fasman算法),这些数据百度都很容易找到。
然后是窗口大小的选择。经过测试,隐层节点数为1024,当窗口大小达到79以上时,线下MaF达到饱和,为0.749。再调节隐层大小为2048,最后的线下MaF为0.767。
该模型提交后线上结果为0.7312。(滑窗模型其实等价于基于整条序列的CNN模型)
Word2vec+深层NN
NN的结构设计主要参考论文《Protein Secondary Structure Prediction Using Cascaded Convolutional and Recurrent Neural Networks》,这是一篇使用深度学习进行蛋白质二级结构预测的经典论文,文中使用了CNN+BiGRU的结构进行蛋白质二级结构预测,模型结构如下:
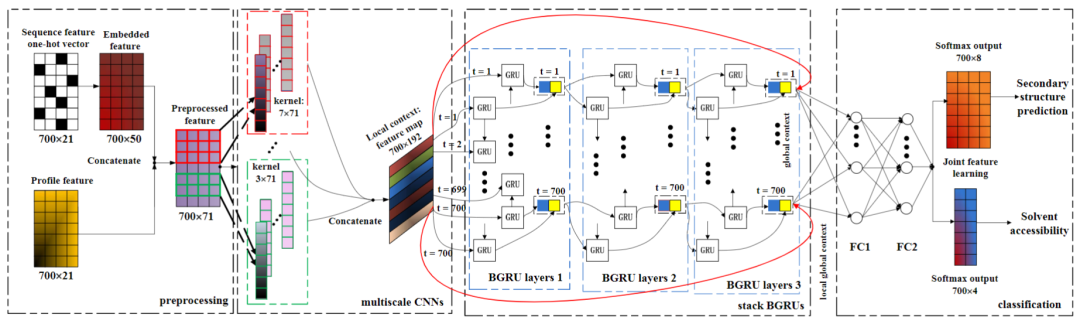
该模型先通过CNN捕获局部信息,再通过RNN融入全局信息,是NLP长文本任务的常见baseline模型。这里基本照搬了模型结构,但将编码部分改为了word2vec预训练的结构,词向量大小为128,其它结构和参数与原文一致,文章可从github项目目录进行下载。
此处还需注意一定的是,如果embedding层是单独对每个氨基酸进行编码的话,那么词表大小为23(数据集中共23种字母)。而在NLP种经常用到的一种叫做n-gram的技术,即将多个词绑定在以此形成整体,这个技术在蛋白质序列种也用得比较多,成为k-mers。
倘若使用k-mers构建词表的话,假定k=3,那么词表的大小就是232323=12167,这样相当于在编码时将上下文也考虑了进去,增加了词的多样性,在一定程度上可以提高模型的学习能力,但也会增大过拟合的风险。
这里我也分别尝试了k=1和k=3的两个模型情况,线下分别为0.719和0.706,线上分别为0.7576和0.7518。
这两个模型的输入和数据划分都有较大差异,显然会有一定的融合收益,将二者的结果进行加权平均后,线上结果为0.7702。
最终模型
将以上几个模型的特征输入都有着较大不同,进行简单加权融合后,线上结果到达0.7770。
在得到以上结果后,进一步分析问题:
模型真正学会的到底是什么:模型主要是记忆了大量由X->Y的固定映射或搭配,然后根据不同搭配的置信度进行决策,学会如何权衡不同搭配以得到更加正确的结果,这也是选用单层小窗口CNN时严重欠拟合的原因。
氨基酸的编码表示:蛋白质总共包括的氨基酸种类较少,在本数据中只有23种,只需要一个23维的onehot向量就可以表示,这也是简单的onehot编码+大窗口CNN能如此有效的原因,也验证了前面的观点,即模型主要是记忆了大量由X->Y的隐射,预测时根据所记忆的大量先验知识,对输出进行决策。
综上,我们设计了最终的模型:在3.2部分的模型中,将embedding部分改成了25维onehot编码+14维理化特征+25维word2vec特征,其中onehot和理化特征部分在训练过程中是frozen的,而word2vec会随着训练进行finetune;其次是加大了CNN部分的窗口,设置成了[1,9,81]。
最终按次方案训练了一个3折的模型,线下MaF平均为0.756。将3折的模型加权平均后线上分数为0.7832。
Top2方案分享
分享原文:https://tianchi.aliyun.com/notebook-ai/detail?postId=97920
建模思路
由于氨基酸序列和二级结构序列的长度相等,我们考虑将序列中的某个氨基酸作为样本 ,其对应位置的二级结构作为标签 ,作为多分类任务。下面部分的代码进行数据预处理,将原始数据转换为单个氨基酸的样本,并保留了序列顺序,以id标记属于统一序列的氨基酸。
特征工程
作为树模型中非常重要的一部分,我们考虑了这些特征:
首先将氨基酸和二级序列编码为连续的数值型变量(label encode),以便输入模型。 除了当前位置的氨基酸外,构造前后各20个氨基酸的shift特征,并都作为categorical feature输入模型。 将当前氨基酸所属蛋白质序列含有的各种氨基酸数目和占比作为特征。 将当前位置的氨基酸及其前后各10个氨基酸作为窗口,统计各类氨基酸的数目。
以下的特征尝试过,但收效甚微,后续不予考虑:
氨基酸的PseAA特征(一些理化属性)及相关特征。 氨基酸word2vec后的embedding向量
模型训练及tricks
首先用了6折交叉验证(cv)进行训练,每折大概450轮左右早停。然而docker提交运行时间限制30分钟,测试集上预测如果使用交叉验证的模型,预测时间完全不够。
考虑使用全部训练集进行训练,手动调试迭代轮数。提交时发现全数据训练的模型大概使用580轮为最佳,30分钟可以运行两个全数据训练模型,因此使用了两个全数据训练模型的融合。
Top3方案分享
分享原文:https://tianchi.aliyun.com/notebook-ai/detail?postId=98092
开源代码:https://github.com/yjh126yjh/TianChi_Protein-Secondary-Structure-Prediction
特征选择与处理
由于自然界中只存在20种常见氨基酸,题目中出现23种,有2种特别少,我们按照生物学对氨基酸的分类标准将其归入X,代表未知氨基酸。
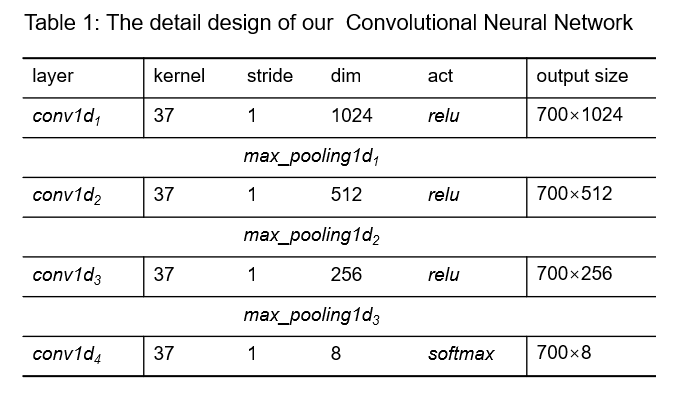
对于一个一级结构的字母,我们选择38维的特征向量来表示它,其中前21维为21种氨基酸的onehot,考虑到氨基酸侧链性质对结构影响较大,我们设置后17维为氨基酸的理化性质onehot,这里理化性质主要包括侧链残基的类别,带电性,极性,亲疏水性,相对分子量等,具体数据来源于Table of standard amino acid abbreviations and properties。
此外,为了便于处理,我们统一将不同长度的氨基酸序列padding到700,保证比目前已知的最长氨基酸序列长,因此20000条氨基酸序列处理后可得到(20000 × 700 × 38)。
模型介绍与训练
我们使用了一个朴素的四层卷积神经网络模型,每层的kernel_size均选择37,以一个较大的局部感受野模拟氨基酸序列的长程作用,使用较高维度的输出通道学习更多更复杂的特征,同时使用dropout和pooling防止过拟合。我们使用95%的数据用于训练,5%的数据用于验证,网络训练时的参数如下:
-learning_rate: 0.00005
-drop_out: 0.4
-batch_dim: 128
-nn_epochs: 300
-loss: 'categorical_crossentropy'
-optimizers: Adam
在我们四块2080Ti的机器上,大概40s一个epoch,然后大概在280 epoch的时候有最佳结果,最佳score对应的模型参数及测试的代码均上传GitHub。
Top5方案分享
分享原文:https://tianchi.aliyun.com/notebook-ai/detail?postId=98009
本方案将赛题建模为一维的语义分割,主要看重语义分割模型能够实现“像素点”的分类。采用的特征有:
氨基酸代号onehot 氨基酸理化特征:疏水值,带电性,分子量,pI,族类,Pa,Pb,Pt,Fi,Fi1,Fi2,Fi3,Pe
由于本人非生化相关专业,故仅仅是从论文、百科中搜集到特征值并直接使用,并未作进一步分析。
特征构造
经分析,题目中的氨基酸序列除了常见的20种氨基酸外,还有3种氨基酸。由于仅查到了20种常见氨基酸的特征数据,故对这3种氨基酸的特征分别采用-1、-2、-3填充。
数值特征归一化到[0,1]区间,“族类”特征采用onehot编码。对序列进行padding使长度一致,padding的部分采用新的onehot编码以示区分,特征值填充为全0。
网络构造
网络构造参考了EfficientDet中的BiFPN结构,大幅简化了backbone层数。经测试kernel size稍大些效果较好。
focal_loss效果不佳, mobile block拖慢了训练速度,实际上没有使用。这里为了程序的完整性予以保留。
往期精彩回顾
本站qq群851320808,加入微信群请扫码: