
世界上最大的量子化学数据集,助力新材料设计和药物发现
电子波函数计算是计算量子化学的一项基本任务。波函数参数的知识允许人们计算分子和材料的物理和化学性质。不幸的是,即使对于简单的分子,解析地计算波函数也是不可行的。
Hartree–Fock 方法或密度泛函理论 (DFT) 等经典量子化学方法允许计算波函数的近似值,但计算量非常大。降低计算复杂性的一种方法是使用能够以低得多的计算成本提供足够好的近似值的机器学习模型。
在最新的一项工作中,俄罗斯 AIRI 的研究人员介绍了一个新的精选类药物分子电子结构的大规模数据集,同时为多分子环境中分子特性的估计建立一个新的基准,并使用该基准评估各种方法。
研究表明,当从单分子设置切换到多分子设置时,最近开发的机器学习模型的准确性会显著下降。而且,这些模型缺乏对不同化学课程的概括。此外,这项工作提供的实验证据表明,更大的数据集会在量子化学领域产生更好的 ML 模型。
该研究以「nablaDFT: Large-Scale Conformational Energy and Hamiltonian Prediction benchmark and dataset」为题,于 2022 年 10 月 24 日发布在《Physical Chemistry Chemical Physics》。

电子的多粒子薛定谔方程(SE)的解使得在分子的化学键水平和晶体的能带结构水平上描述物质成为可能。同样,物质的电子系统决定了它的大量平衡和传输特性,这为寻找新分子(如有前途的药物或催化剂)和新材料(如新型超硬、超导、低维和其他材料)开辟了巨大的机会。
解决多粒子 SE 是一项复杂的任务,吸引了几代研究人员的大量关注,但不幸的是,其解析解仍然未知。然而,存在各种各样的数值方法可以在不同的精度水平上解决它。这些方法包括一个层次结构,该层次结构在准确性与计算成本以及使用特定技术可以在合理时间内计算其运动的电子数量之间进行权衡。
在层次金字塔的顶端是两类 Post-Hartree–Fock 方法和量子 Monte Carlo 方法。它们非常准确(大约 1 kcal mol^-1)但计算量大,允许考虑多达数十个电子的系统。所有这些都是基于操纵多体波函数,多体波函数表示为具有可调系数的单电子轨道的展开。在这些可调系数的空间中执行优化搜索,以找到提供系统最小能量的多粒子波函数。因此,它最接近基态(最小能态)的“真实”多粒子波函数。
层次结构的第二步采用密度泛函理论(DFT)方法,这是目前解决电子多粒子 SE 的主要方法。
DFT是一种平均场法,将多粒子问题分解成若干个单粒子问题,一个电子在其他电子的有效场中求解SE。这种方法与更准确的方法之间的主要区别在于它操纵的不是多粒子波函数,而是电子密度,这是一个可观察的量。DFT 可以以令人满意的精度(大约 10 kcal mol^-1 )考虑 1000 个电子规模的系统,从而扩大到已经是纳米物体的系统,例如纳米管和漂白剂、蛋白质片段或催化表面的一部分。
DFT 的准确性由所谓的交换相关 (XC) 泛函决定,它本身也有一个准确性/复杂性权衡层次。人们相信,通过寻找快速准确的交换相关泛函,有可能将 DFT 的精度提高到 1 kcal mol^-1,从而使其在精度上几乎与顶级方法相当。
在层次结构的第三步是所谓的参数方法,例如紧束缚方法,它需要哈密顿量的参数化。它们使计算多达数万个电子的广泛系统成为可能。然而,不确定的预参数化步骤和所得精度的较大波动性使得该方法不如 DFT 受欢迎。
除了用于解决电子多体 SE 的传统数值方法外,机器学习 (ML) 方法也大量涌现,在准确性/复杂性的层次结构中寻找自己的位置。
将 ML 纳入该领域的一个有前途的方向是开发一系列基于深度神经网络 (NN) 的试验波函数;近期的研究结果表明,它可以胜过最好的高精度量子 Monte Carlo 方法。另一个方向是从原子坐标(系统配置)直接预测波函数、电子密度或哈密顿矩阵。第三个方向是使用神经网络对 XC 泛函建模以实现高精度 DFT。
从头计算分子性质预测的一般框架包括两个步骤:首先计算特定分子构象或一组构象的电子结构,然后根据第一步的结果计算所需的性质。第二步相对简单,但总的计算复杂度可能太高,具体取决于第一步使用的方法。
避免这种复杂性的一种直接方法是训练机器学习模型以直接预测所需的分子特性,绕过电子结构部分。然而,这种方法可能缺乏泛化性,因为需要为每个新属性开发和训练一个单独的新模型。
最近的研究表明,使用多种不同的 ML 方法在电子结构预测领域取得了可喜的成果。它通过用相对简单的 ML 模型代替它来避免 DFT(或高阶)方法的昂贵计算,但保留广义属性计算框架。这样,该方法只需要一个 ML 模型来满足所有必要的属性。
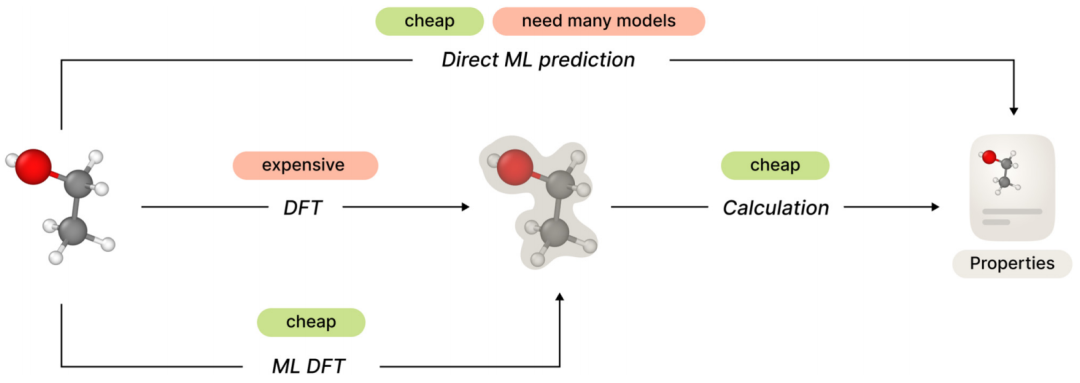
尽管最近在使用 ML 的哈密顿矩阵近似方面取得了进展,但这些研究存在两个严重的缺点。首先,所有模型都在单分子设置中进行了训练和测试(对同一分子的不同构象进行训练和测试);其次,所有模型都存在扩展到更大分子结构的问题。
除非得到训练数据的大小和可变性的支持,否则机器学习模型的表达能力毫无意义。相关领域正在见证小分子和化合物的大规模数据集的兴起,这些小分子和化合物的必要特性已通过准确且计算成本高昂的方法建立;例如,MOSES 基准测试平台基于 ZINC clean leads 数据集的一个子集比较了药物发现的分子生成模型。具有 DFT 计算结果的大规模数据集的其他示例是 Open Catalyst 2020 (OC20) 和 2022 (OC22)。这些数据集总共包含 130 万个分子松弛,以及超过 2.6 亿次 DFT 计算的结果。
大规模数据集使自然语言处理领域取得了令人瞩目的成果。基于 Transformer 的模型(例如 BERT 或 GPT-3)成功的关键原因之一是可以访问庞大的训练语料库。在药物化学领域已经表明,从完整词典到词典的 30% 的准确性下降对于临床试验中的疾病链接具有重要意义。除了质量提高之外,更大、更多样化的数据集对于模型的稳健性也很重要。之前,已经有团队阐述了机器学习模型的泛化能力受测试实体/关系是否已出现在训练集中的影响。
nablaDFT 数据集
俄罗斯 AIRI 的 DL in Life Sciences 研究小组介绍了一个新的大型数据集 nablaDFT,其中包含约 100 万个分子结构的约 600 万个构象的约 100 万个构象的结构和哈密顿矩阵,具有使用 Kohn-Sham 方法计算的电子特性。该数据集允许在不同设置下比较基于 DFT 的模型,特别是训练集和测试集包含不同分子的泛化测试。
在基准测试方面,该团队采用了几种经典的和最先进的基于 DFT 的模型,并在其数据集上比较了它们的结果,得出了关于它们的表现力、泛化能力以及对数据大小和训练制度的敏感性的重要结论。这项工作中考虑的模型有两种,一种是估计势能估计,另一种是预测哈密顿系数。
通过 GitHub 提供的数据集包含超过 100 万种药物样分子的超过 500 万种构象,以及构象能量、DFT 哈密顿矩阵、波函数等量子特性。单个构象计算平均需要大约 5 分钟的 CPU 时间,整个数据集总共需要大约 50 年的 CPU 时间。
基准测试结果
为了在不同设置中对模型进行基准测试,研究人员将测试集分为三个子集:
训练集中呈现的结构的分子构象。
训练集中未出现的结构的分子构象。
训练集中未呈现具有支架的结构的分子构象。
所有模型都在多分子设置中进行了训练。根据实验结果,最佳模型在构象能量预测任务的分离结构测试集上实现了 3.2x10-2 hartrees (~20 kcal/mol) 的平均误差,而湿实验室可达到的化学准确度约为 1kcal/mol。毫不奇怪,大多数模型在对已见分子结构的新构象进行测试时表现更好。即使是简单的线性回归模型也显示出从 4.7x10^-2 Hartree MAE 到 4.0x10^-2 hartrees 的改进。
尽管获得接近化学准确性的模型仍然是一个挑战,但该实验证据表明,更大的数据集会产生更好的 ML 模型。
论文链接:https://pubs.rsc.org/en/content/articlelanding/2022/CP/D2CP03966D
相关报道:https://phys.org/news/2022-11-world-largest-quantum-chemistry-dataset.html
人工智能 × [ 生物 神经科学 数学 物理 材料 ]
「ScienceAI」关注人工智能与其他前沿技术及基础科学的交叉研究与融合发展。
欢迎关注标星,并点击右下角点赞和在看。
点击阅读原文,加入专业从业者社区,以获得更多交流合作机会及服务。