
刷新认知!基因表达,男女有别|Science、Cell等发布15篇人类遗传调控新成果
生物信息学习的正确姿势
NGS系列文章包括NGS基础、在线绘图、转录组分析 (Nature重磅综述|关于RNA-seq你想知道的全在这)、ChIP-seq分析 (ChIP-seq基本分析流程)、单细胞测序分析 (重磅综述:三万字长文读懂单细胞RNA测序分析的最佳实践教程)、DNA甲基化分析、重测序分析、GEO数据挖掘(典型医学设计实验GEO数据分析 (step-by-step))、批次效应处理等内容。

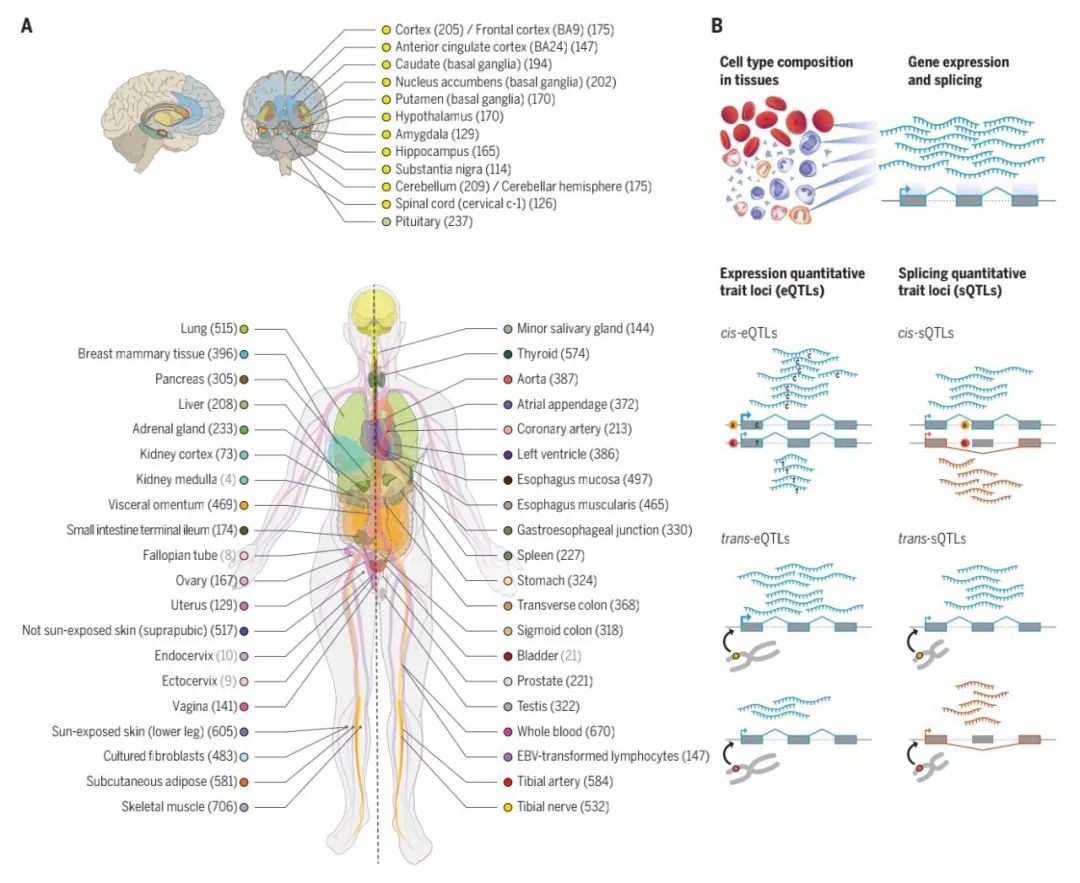
图:GTEx v8研究样本及数据类型

图:GTEx最新研究总览
附15项研究成果速览


题目:GTEx项目构建人体组织遗传调控效应图谱
研究团队介绍了GTEx(v8)版本数据的分析,检测了来自838个死后捐赠者49个组织的15201个RNA测序样本。该研究全面表征了顺式和反式基因表达和剪接的遗传关联,表明几乎所有基因都存在调节关联,并描述了潜在的分子机制及其对等位基因异质性和复杂性状的多效性的贡献。
文章链接:
https://science.sciencemag.org/content/369/6509/1318.abstract
题目:性别对跨人体组织基因表达的影响
题目:细胞类型特异性基因调控基因在人体组织中的表达
题目:端粒长度在人体组织中的决定性作用
Determinants of telomere length across human tissues
研究团队表征了来自6391个组织样本端粒长度的变异性,这些样本代表了GTEx项目的20多个组织类型和952个个体。研究描述了不同组织类型之间的差异,以及组织类型之间的正相关以及与年龄和血统的关联,发现遗传变异可影响多种组织类型中的端粒长度,并且端粒长度可能介导年龄对基因表达的影响。
文章链接:
https://science.sciencemag.org/content/369/6509/eaaz6876
题目:跨人体组织的转录特征可识别功能性罕见遗传变异
Transcriptomic signatures across human tissues identify functional rare genetic variation
确定稀有遗传变异的功能和表型影响是一项重大挑战。研究团队通过分析基因表达、等位基因特异性表达和多组织RNA测序数据的可变剪接,扩展了基因驱动的转录组异常检测,并证明了每种信号都可以指导稀有变异的独特分类。该研究结果将成千上万的稀有变异链接到各种分子效应,为将稀有变异影响转录组与人类特征相关联提供了证据。
文章链接:
https://science.sciencemag.org/content/369/6509/eaaz5900
题目:组织特异性遗传特征为临床试验中药物副作用的预测提供依据
Tissue-specific genetic features inform prediction of drug side effects in clinical trials
通过结合48个组织中的基因表达和eQTL,该研究评估了> 360000个英国生物库个体中1167个表型的全表型关联研究(PheWAS),以分析是否可在临床试验中预测药物副作用。研究确定了具有五个遗传特征的药物靶基因,包括基因表达的组织特异性。与没有这种特征的基因相比,其带来的副作用风险增加了2.6倍。该研究展示了来自多个组织的PheWAS和eQTL数据用于药物副作用预测的效用,并强调了组织特异性药物递送的需求。
文章链接:
https://advances.sciencemag.org/content/6/37/eabb6242
题目:PhenomeXcan:通过转录组将基因组映射到表型
PhenomeXcan: Mapping the genome to the phenome through the transcriptome
PhenomeXcan是将来自GTEx(v8)49个组织的4091个性状转录组数据与GWAS研究统计数据中887万个变体整合的一个可查询基因平台,其中包括22515个基因。研究团队开发了一种新颖的贝叶斯共定位方法,即快速富集估计辅助共定位分析(fastENLOC),可先考虑可能的因果基因-性状关联。利用PhenomeXcan,研究团队提供了新的和未报告的基因与表型关联以及复杂的基因特征簇。PhenomeXcan(phenomexcan.org)为转录研究提供了对复杂数据的广泛及用户友好的访问。
文章链接:
https://advances.sciencemag.org/content/6/37/eaba2083
题目:人体定量蛋白质组图谱
A Quantitative Proteome Map of the Human Body
研究团队从32个正常人体组织中超过12000个基因中定量了相对蛋白质水平。通过鉴定组织特异性或组织富集的蛋白质,并将其与转录组数据进行比较,许多普遍存在的转录本被发现可编码组织特异性蛋白质。RNA和蛋白质富集的差异揭示了分泌蛋白合成和作用的潜在位点。此外,该研究表明蛋白质组织富集信息可以解释遗传疾病的表型,而仅靠转录本信息是无法获得的。
文章链接:
https://www.cell.com/cell/fulltext/S0092-8674(20)31078-3
题目:Primo:整合多个GWAS和omics QTL汇总统计数据,阐明与性状相关SNP的分子机制并检测复杂性状的多效性
Primo: integration of multiple GWAS and omics QTL summary statistics for elucidation of molecular mechanisms of trait-associated SNPs and detection of pleiotropy in complex traits
为全面解释已知的性状相关SNP如何影响复杂性状,研究团队提出了一种Primo方法,用于对来自不同细胞条件或研究的系列omics QTL汇总统计数据进行GWAS统计数据的综合分析。Primo方法可研究SNP与复杂和组学特征的关联模式。在含有已知易感位点的基因区域,Primo可进行条件关联分析,以解释连锁不平衡问题。Primo允许进行未知的异质性和样本相关性研究。
文章链接:
https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-02125-w#auth-Lin_S_-Chen
题目:sn-spMF:基质分解告知组织特异性基因表达的遗传调控
sn-spMF: matrix factorization informs tissue-specific genetic regulation of gene expression
研究团队开发了一个受约束的矩阵分解模型sn-spMF,以学习组织共享的模式将其应用于GTEx项目的49个人体组织。学习因子(learned factors)可反映具有已知生物学相似性的组织,并识别可能介导组织特异性作用的转录因子。sn-spMF可在https://github.com/heyuan7676/ts_eQTLs获取。
文章链接:
https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-02129-6
题目:大量跨人体组织等位基因的表达数据
A vast resource of allelic expression data spanning human tissues
研究团队介绍并演示了从GTEx(v8)版本中生成的大量等位基因表达资源的实用性,其中包含15253个样本,覆盖54个人体组织,SNP级别的等位基因总计为4.31亿,单倍型水平的总量为1.53亿。此外,研究团队扩展了phASER工具,允许使用单倍型水平的等位基因数据估算顺式调节变体的效应大小。这是迄今为止最大的等位基因资源,且能够公开提供单倍型水平的等位基因数据。
文章链接:
https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-02122-z
题目:GTEx中外源和祖源对eQTL分析和GWAS共定位的影响
Impact of admixture and ancestry on eQTL analysis and GWAS colocalization in GTEx
研究团队在GTEx(v8)中识别了117个具有高度群体混合个体的子集,并估计了全基因组的局部祖源信息。在七个组织中使用混合样本进行全基因组顺-eQTL定位,并通过祖源信息进行调整。最后,该研究确定了与本地祖先高度相关的一部分eQTL变体。为GTEx(V8)版本中的混合个体提供了本地祖先图,并描述了祖先和混合物对基因表达,eQTL和GWAS共定位的影响。
文章链接:
https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-02113-0
题目:PTWAS:利用TWAS概率分析研究组织相关复杂性状的因果分子机制
PTWAS: investigating tissue-relevant causal molecular mechanisms of complex traits using probabilistic TWAS analysis
研究团队提出了一种新的计算框架,即概率全转录组关联研究(PTWAS),以研究基因表达与复杂性状之间的因果关系。PTWAS应用工具变量分析的既定原则,利用概率eQTL注释来描述和解决TWAS中出现的独特挑战。PTWAS不仅具有比现有方法更高的功能,而且还提供了新颖的功能来评估因果关系假设,以及评估组织或细胞类型特异性基因对性状的影响。研究团队通过分析来自GTEx(v8)49个组织的eQTL数据和114个复杂性状的GWAS统计数据证明了PTWAS的强大功能。
文章链接:
https://genomebiology.biomedcentral.com/articles/10.1186/s13059-020-02026-y
题目:精细映射和QTL组织共享信息提高了因果基因识别的可靠性
Fine‐mapping and QTL tissue‐sharing information improves the reliability of causal gene identification
近年来,通过转录表达整合转录组学研究和GWAS研究已得到广泛应用,使得GWAS基因座的功能表征和因果基因的预测成为可能。但最佳的预测性能模型不一定会导致更可靠的因果基因发现。为在不增加假阳性的情况下改善目标基因的发现,研究团队使用GTEx项目中948个供体54个组织的17382个RNA测序样本的表达和剪接数据,开发并比较了多种转录组预测方法。研究发现,通过精细映射(dap-g)和跨组织借用信息(masher)来反映具有因果概率的预测模型可以在重要关联的数量和比例方面提供更好的性能。所有的预测模型都可以在predictdb.org上公开获得。
文章链接:https://onlinelibrary.wiley.com/doi/full/10.1002/gepi.22346
题目:用于检测转录物变体调控作用的多克隆等位基因表达测定方法
A polyclonal allelic expression assay for detecting regulatory effects of transcript variants
研究团队提出了一种利用CRISPR / Cas9检测基因变异在转录组中调节作用的实验方法,然后进行靶向测序。利用该检测方法,研究团队对整个基因组和两个孟德尔遗传病基因中的32个提前终止变异体,HEK293T细胞中的33个eQTL预测因果变异体和62个对照变异体进行了分析,并复制HeLa细胞中的一部分变异体。结果显示,该方法能够捕获eQTL变体以及提前终止变异体触发的无意义介导衰变的调节作用,表明该方法可用于验证遗传变异的转录组水平效应。
文章链接:
https://genomemedicine.biomedcentral.com/articles/10.1186/s13073-020-00777-8#article-info
参考资料:
1. ‘Invaluable’ database helps solve mystery of how genes are regulated
https://www.sciencemag.org/news/2020/09/invaluable-database-helps-solve-mystery-how-genes-are-regulated
2. GTEx Studies Provide Comprehensive Map of Genetic Regulatory Variation Across Cell Types, Tissues
https://www.genomeweb.com/genetic-research/gtex-studies-provide-comprehensive-map-genetic-regulatory-variation-across-cell#.X1rSiHot1PY
3. https://commonfund.nih.gov/GTex
4. https://commonfund.nih.gov/gtex
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|



























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集
喜欢别忘了点“在看”呦!