
图神经网络在生化医疗方面的相关应用

图神经网络之所以得到如此广泛的关注,得益于它的应用场景丰富。
本文就来分享一下图神经网络在生化医疗方面的相关应用。
以下内容节选自《图神经网络:基础与前沿》一书!

--正文--
▊ 预测分子的化学性质和化学反应
在生化领域,药物分子化合物、蛋白质等经常被作为研究对象。以分子为例,它是一个天然的图结构,可以将分子中的原子看作节点,将化学键看作边,研究分子的化学性质就可以看成给一个图分类或者回归问题。事实上,在机器学习图分类问题的标准数据集中,生化分子占据了非常大的比例:对于化合物来说,MUTAG 数据集旨在分类它们是否为芳香剂,Tox21 数据集分类不同的毒性,NCI-1 分类对癌症的阻碍作用。对于这类问题,我们一般通过学习整个分子图的表示得到所谓的“分子指纹”,然后用它做各种性质的预测。
分子指纹的学习和化学性质的预测在图神经网络发展的早期起到了非常重要的作用。例如,Duvenaud 等人在传统的分子指纹的基础上做了改进,发展了卷积的方法,也是早期图卷积网络的一种;而Glimer 等人提出的消息传递网络更是在分子化学的背景下对所有信息传递网络做了统一和改进。除此之外,图卷积网络还被用来预测两个分子的化学反应或者寻找有效的抗体。由于前面的章节已经对图分类任务做了介绍,本节就不赘述了。本节简单介绍化学反应预测这个比较新颖的应用。
化学反应预测是指给定一些反应物分子图

(图神经网络用于化学反应预测的过程)
▊ 图生成模型与药物发现
药物开发是一个耗时、费力的大工程,从最初的药物设计、分子筛选,到后期的安全测试、临床试验,一般会花超过10 年的时间而且不能保证成功率。因此,面对COVID-19 这种突发的流行病,我们无法立刻研发出有效药物,而只能在已知的药物里寻找可能有效的进行药物重用。为了加快新药开发的进程,人工智能在药物发现领域起到了越来越重要的作用,尤其是在新分子的设计阶段,图神经网络与图生成模型的应用极大地提升了药物发现的效率。虽然分子也可以表征成SMILES 字符串的形式(如下图所示),但是我们很难从这种字符串中直接获得语法和结构信息,因此更常用的分子生成方法是把分子当成图来生成。

(分子的图表示与对应的SMILES 字符串表示)
深度学习在解决生成问题的能力上早已声名远扬,生成对抗网络(Generative Adversarial Networks,GAN)和变分自编码器等深度生成模型被广泛应用于图像和文本生成领域,然而把这些模型扩展到分子图的生成问题上并不容易。
首先,由于分子具有不同类型的节点和不同类型的边,导致一个很小的分子也有着很大的搜索空间;其次,由于图的不规则性,设计一个解码器从一个向量生成一个图结构是非常有挑战的;最后,我们还需要保证生成的图是一个分子,并且具有我们想要的化学性质,这就要求生成过程中有很多的限制条件。一般来说,图生成模型有以下几类:自回归(Auto-regressive)模型,基于生成对抗网络的模型,基于变分自编码器的模型,以及基于标准化流(Normalizing Flow)的模型。
首先,我们对分子图生成问题给出一个形式化的定义:
定义5 给定一些已知的分子图
和它们对应的化学性质 ,假设它们都服从某种未知的分布p(G),图生成模型的目标是学到这个分布并从中采样出新的分子图 。这些生成的新分子图需要是有效的分子(满足化学价等条件),并且具有我们想要的化学性质。
1. 自回归模型
GraphRNN 是一个早期的经典图生成模型,它把图的生成过程看作一个序列生成过程。虽然图结构中节点的位置是可互换的,但是生成图总要有个先后顺序,所以GraphRNN 中每个图的节点被预先设定了一种排序,按照这种排序,我们可以一个节点一个节点地生成这个图。在下图中,我们每次生成一个节点,然后把这个节点连接到已经生成的其他节点上,也就是生成节点所对应的边,这样依次循环,直到最终生成整个图。

(GraphRNN:图生成的自回归模型)
具体来讲,我们先把图G 用宽度优先搜索表示成一个序列的形式:
其中,
这里,
GraphRNN 是一个比较通用的图生成模型,它没有考虑分子的性质,所以在分子的生成问题上,可以用强化学习的方法加上对生成分子的化学性质的预测,作为奖励函数进行反馈,从而使生成的分子具有我们想要的化学性质。
2. 基于生成对抗网络的模型
生成对抗网络由两部分组成:生成器
生成对抗网络的出现,引领了图像生成等任务的热潮,然而在图数据上,同样的难题摆在图神经网络面前:怎么设计解码器。下图给出了一个最简单的例子,这个模型叫作MolGAN。首先,它把分子图表示成两个部分:邻接张量(比邻接矩阵多了一个维度,用来表示边的类型)和节点属性矩阵。它的解码生成过程分为两个分支,从一个采样的图向量
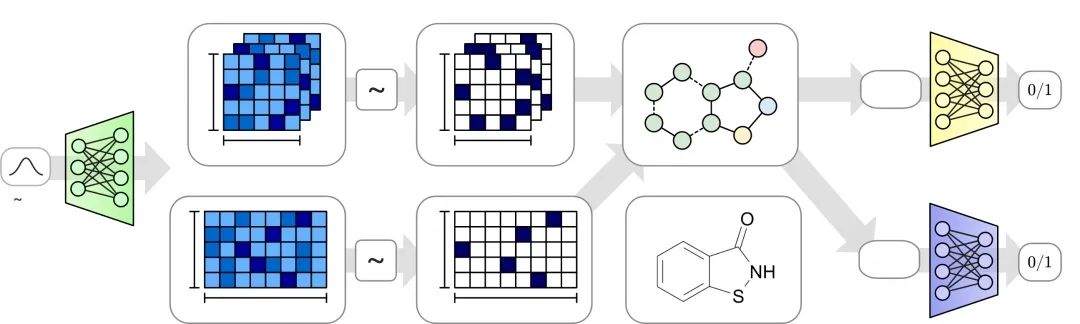
(MolGAN [130]:图生成的图神经网络模型)
注:N 为节点(原子)数量,T 为边(化学键)的模型的数量
3. 基于变分自编码器的模型
变分自编码器由一个编码器把样本
其中,后验概率
《图神经网络:基础与前沿》一书的5.3.1 节介绍了图变分自编码器,但这个模型只是用来作为图的无监督表示学习或者链路预测的,因为它的编码器得到的结果是关于节点的表示。如果要生成新的图,在解码时我们不知道有多少节点及这些节点是怎么连接的,所以只能从一个表示整个图的向量
一个比较经典的用于分子图生成的变分自编码器模型是联结树VAE(Junction-Tree VAE,JT-VAE)(如下图所示),它的思想是:如果按照自回归模型中一个节点一个节点依序生成,很难保证生成的分子图是符合语法的,那么不如先把图分解成一些子结构,再拼接这些在一定语义中有效的子结构。首先,我们把分子图

(JT-VAE:基于联结树的VAE 生成分子图的过程)
除了采用子结构来保证分子图的语义有效性,另一些基于变分自编码器框架的图生成模型则侧重于增加图解码器的限制。例如,Liu 等人在生成图的边时采用遮蔽(mask)技术保证去除违反分子语义结构限制的边。具体来说,在生成边的概率分布上增加一个变量
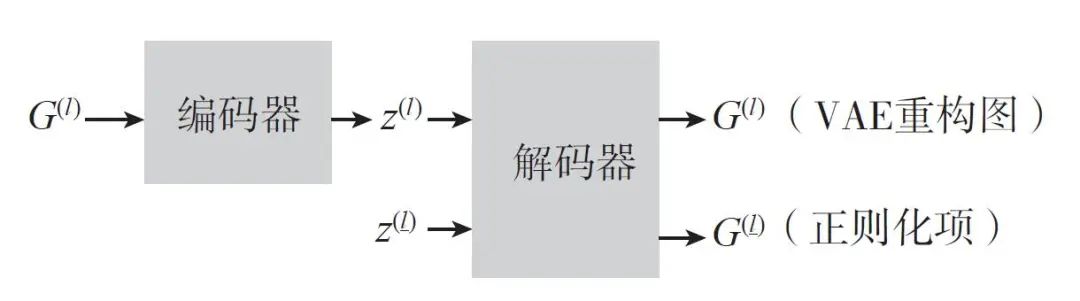
(增加限制的变分自编码器。通过采样近似之后,我们在原来的变分自编码器的基础上增加另一个分支,采用一个新的向量
4. 基于标准化流的模型
标准化流是一种比较新的深度学习生成模型,它的主要思想是使生成过程可逆,这样从隐藏向量到生成样本的过程就不会损失信息。我们将其与变分自编码器进行简单的对比,在变分自编码器中,我们需要一个编码器
实现
其中,
然后,在下一层对
GraphNVP(如下图所示)就是一个把Real NVP 应用到图生成任务上的例子,它分别对分子图的邻接张量和节点属性矩阵应用仿射耦合层,得到一个代表邻接张量的向量
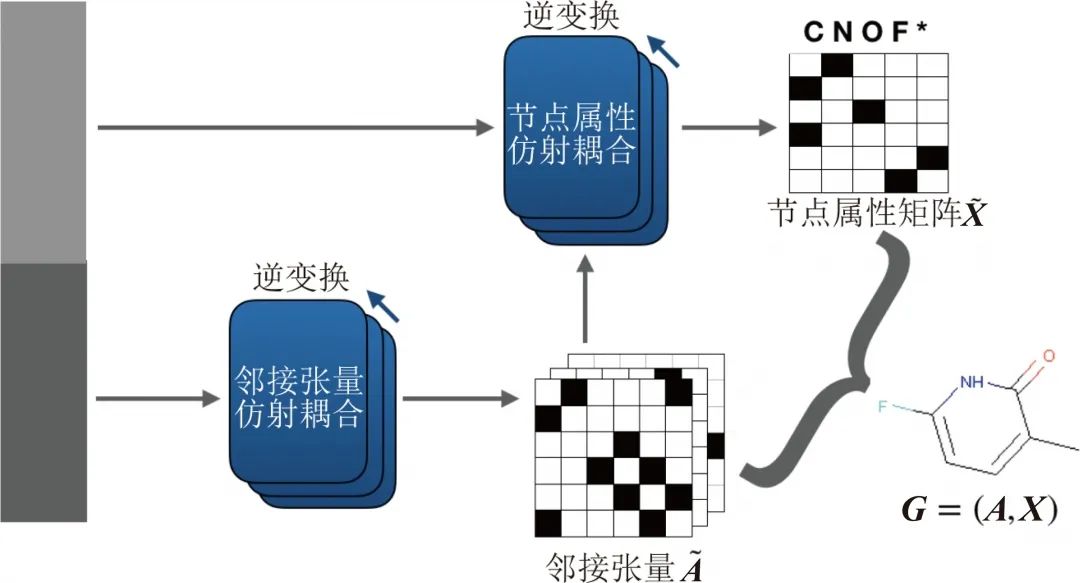
(GraphNVP:图生成的标准流模型)
▊ 药物/蛋白质交互图的利用
除了分子本身的化学结构可以被认为是图,在生化领域,另一种重要的图是蛋白质交互图。虽然蛋白质本身有时也被认为是图,但是更普遍的情况是将蛋白质作为节点,将蛋白质之间的相互作用当作边,组成一种蛋白质交互图。
蛋白质交互图也是图神经网络常用的一个标准数据集,它主要用来做(归纳式学习的)节点分类预测(如GraphSAGE)。类似的还有DDI 图和DTI (Drug-Target-Interaction)图,DDI 用来研究药物之间的相互作用(可以是正的协同作用,也可以是不良反应),而DTI 主要用在研究药物和目标蛋白质之间的相互作用,它们都在药物研发中起到很重要的作用。由于在药物开发中,我们对药物成分的研究总是不完全的,DDI 图和DTI 图上的研究集中在通过已知的图结构预测那些未知的相互作用,也就是链路预测的问题。通过对DDI 的预测,可以防止推荐药物时可能产生的药物之间的不良反应,而DTI 的预测则能帮助我们理解药物机制及将旧药新用。对于链路预测这个经典问题,我们不再做更多介绍,下面我们来看图神经网络是怎么利用DDI 图的信息做更安全的药物推荐的。
基于电子病历的药物推荐是医疗电子化的一个重要方向,但是仅考虑病人历史记录的推荐有一个问题,那就是很有可能会推荐药物相互之间有不良反应的组合,因此,利用DDI 图的信息就是一个必然。GAMENet采用了图神经网络与记忆网络结合的方式来解决这个问题(如下图所示)。首先,DDI 图被一个图神经网络编码,然后这个编码的信息被放在记忆网络的记忆库中,在每个时间点与记忆网络中的其他动态历史记录共同作用,从而在选择药物推荐时避免药物之间的不良反应。
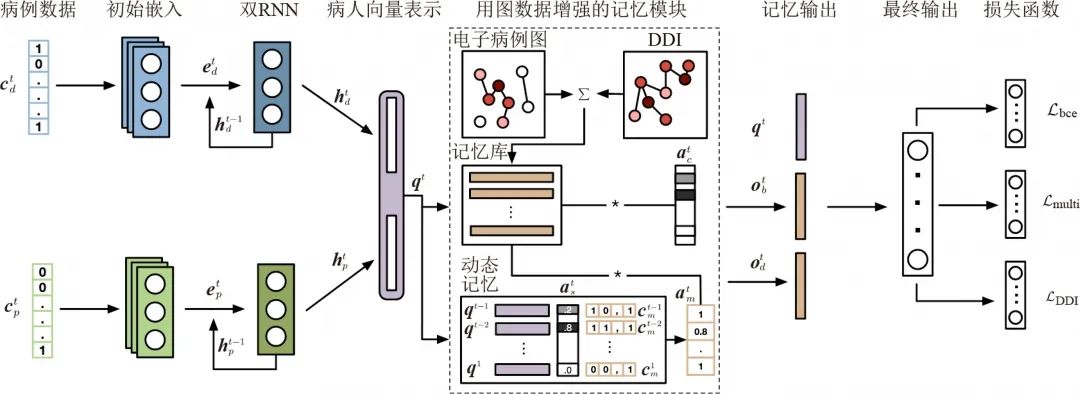
(图神经网络用于药物推荐。DDI 图被图神经网络编码后放在记忆网络的记忆库中,与由电子病历数据所产生的动态记忆共同作用,得到药物的推荐)
▼
参考文献:
[1] DUVENAUD D K, MACLAURIN D, IPARRAGUIRRE J, et al. Convolutional networks on graphs for learning molecular fingerprints[C]//Advances in neural information processing systems, 2015: 2224-2232.
[2] GILMER J, SCHOENHOLZ S S, RILEY P F, et al. Neural message passing for quantum chemistry[C]//Proceedings of the 34th International Conference on Machine Learning-Volume 70. JMLR. org, 2017: 1263-1272.
[3] JIN W, COLEY C, BARZILAY R, et al. Predicting organic reaction outcomes with weisfeiler-lehman network[C]//Advances in Neural Information Processing Systems, 2017: 2607-2616.
[4] COLEY C W, JIN W, ROGERS L, et al. A graph-convolutional neural network model for the prediction of chemical reactivity[J]. Chemical science, 2019, 10(2):370-377.
[5] STOKES J M, YANG K, SWANSON K, et al. A deep learning approach to antibiotic discovery[J]. Cell, 2020, 180(4):688-702.
[6] YOU J, YING R, REN X, et al. Graphrnn: Generating realistic graphs with deep auto-regressive models[C]//International Conference on Machine Learning, 2018: 5708-5717.
[7] YOU J, LIU B, YING Z, et al. Graph convolutional policy network for goaldirected molecular graph generation[C]//Advances in Neural Information Pro-cessing Systems, 2018: 6410-6421.
[8] DE CAO N, THOMAS K. Molgan: An implicit generative model for small molecular graphs[J]. arXiv preprint arXiv:1805.11973, 2018.
[9] JIN W, BARZILAY R, JAAKKOLA T. Junction tree variational autoencoder for molecular graph generation[C]//International Conference on Machine Learning, 2018: 2323-2332.
[10] LIU Q, ALLAMANIS M, BROCKSCHMIDT M, et al. Constrained graph variationalautoencoders for molecule design[C]//Advances in neural information processing systems, 2018: 7795-7804.
[11] MA T, CHEN J, XIAO C. Constrained generation of semantically valid graphs via regularizing variational autoencoders[C]//Advances in Neural Information Processing Systems, 2018: 7113-7124.
[12] MADHAWA K, ISHIGURO K, NAKAGO K, et al. Graphnvp: An invertible flow model for generating molecular graphs[J]. arXiv preprint arXiv:1905. 11600, 2019.
[13] SHANG J, MA T, XIAO C, et al. Pre-training of graph augmented transformers for medication recommendation[J]. arXiv preprint arXiv:1906.00346, 2019.
[14] ZITNIK M, AGRAWAL M, LESKOVEC J. Modeling polypharmacy side effects with graph convolutional networks[J]. Bioinformatics, 2018, 34(13):i457-i466.
[15] MA T, XIAO C, ZHOU J, et al. Drug similarity integration through attentive multi-view graph auto-encoders[C]//Proceedings of the 27th International Joint Conference on Artificial Intelligence. AAAI Press, 2018: 3477-3483.
[16] SHANG J, XIAO C, MA T, et al. Gamenet: Graph augmented memory networks for recommending medication combination[J]. AAAI, 2019.


▊《图神经网络:基础与前沿》
马腾飞 编著
梳理图神经网络(GNN)领域的经典模型
帮助读者构建图神经网络知识体系
厘清重要模型的设计思路和技术细节
展现图神经网络的研究进展
-
图神经网络在推荐系统、生物医疗、自然语言处理等不同场景的实践
图神经网络是人工智能领域的一个新兴方向,它不仅迅速得到了学术界的广泛关注,而且被成功地应用在工业界的多个领域。
本书介绍了图神经网络和图深度学习的基础知识和前沿研究,不仅包括它们的发展历史和经典模型,还包括图神经网络在深层网络、无监督学习、大规模训练、知识图谱推理等方面的前沿研究,以及它们在不同领域(如推荐系统、生化医疗、自然语言处理等)的实际应用。
本书既可作为人工智能领域研究和开发人员的技术参考书,也可作为对图上的深度学习感兴趣的高年级本科生和研究生的入门书。
(扫码了解本书详情)

如果喜欢本文
欢迎 在看丨留言丨分享至朋友圈 三连
热文推荐
▼点击阅读原文,获取本书详情~