外显子和基因组基本概念(一)
基因组(Genome):分子生物学和遗传学领域中指生物体所有遗传物质的总和,包括DNA或RNA(病毒)。DNA具体包含编码DNA、非编码DNA、线粒体DNA和叶绿体DNA。研究基因组的科学称为基因组学。
同源染色体(Homologous Chromosomes):一个物种中形态和结构基本相同的染色体。在二倍体生物细胞中,同源染色体在减数第一次分裂的四分体时期中彼此联会(若是三倍体及其他奇数倍体生物细胞,联会时会发生紊乱),最后分开到不同的生殖细胞中。子代的一对染色体其中的一条来自母方,另一条来自父方。
参考基因组(Reference Genome, REF):又称参考(序列)组装(A Reference Assembly),是一个电子化的核酸序列数据库(A digital nucleic acid sequence database)。它由多个科学家和研究单位协作组装、维护和更新,用以作为一个物种的一个理想化的个体的、全基因组序列的典型代表或案例(但不能保证可以精准地代表某个地球上存在过的生物体)。人类、病毒、细菌、真菌、植物和动物理论上都有各自的参考基因组,目前只有部分物种被测通和公布。NGS基础 - 参考基因组和基因注释文件
人类基因组由23对染色体、约60亿个碱基(或核苷酸)组成。正常人类基因组是以2个拷贝存在(是指同源染色体,而非姐妹染色单体),分别来自父母。人类的基因组有几个不同的版本名,目前比较常用的有hg19、hg38、GRCh37、GRCh38。hg系列是UCSC的叫法,GRCh系列是NCBI和Ensembl的叫法。同一版本的序列是一样的,hg19对应GRCh37,hg38对应GRCh38(坐标与hg19/GRCh37不同)。
参考基因组的实体是一个文本文件(.fasta),通常是个单倍体(除了性染色体),含有染色体号和核酸(A/T/G/C)序列,可压缩与索引,且包含一系列的配套文件(例如:GTF文件,记录每个基因名称及其各种元器件的位置)。参考基因组可提供来自每个供体不同DNA序列的单倍体镶嵌(A haploid mosaic of different DNA sequences from each donor)。
事实上,基因组学、高通量测序以及相关的生信分析技术,很大程度上得益于人类基因组计划(Human Genome Project, HGP)。HGP是一项与曼哈顿原子弹计划和阿波罗计划相提并论的规模宏大、跨国跨学科的科学探索工程。旨在测定组成人类染色体(指单倍体)中所包含的30亿个碱基对组成的核苷酸序列,从而绘制人类基因组图谱,并且辨识其载有的基因及其序列,达到破译人类遗传信息的最终目的。
全基因组测序(Whole Genome Sequencing, WGS):是指利用高通量测序平台对生物的不同个体(或群体)、同一个体的不同器官(或组织、细胞)进行全基因组测序,并进行生物信息学分析(主要是利用统计方法获取影响表型或经济性状的候选基因或功能突变)。
高通量测序(High-Throughput Sequencing, HTS):是对传统Sanger测序(也称为一代测序技术)革命性的改变, 一次(一轮反应或拍照)对几十万到几百万条核酸分子进行序列测定,故又称下一代测序技术(Next Generation Sequencing,NGS)。
高通量测序也被称为深度测序(Deep Sequencing),是人类历史上多学科、基础研究、资本运作与商业化结合的成功案例之一,直接导致了高通量测序仪(当前世界最尖端的大型设备之一)的发明与革新,使得对一个物种的基因组和转录组进行高效、细致、全貌的分析成为常规操作。高通量测序作为分子群体遗传学和个人基因组学研究的有力工具,对21世纪前半叶的生命科学研究、生产、疾病的诊断和治疗起到巨大作用,也对生物信息学的进一步发展起到重要的推动作用。
基因结构:真核生物的基因和基因调控大致分为4个区域,1)编码区,包括外显子与内含子;2)前导区,位于编码区上游,相当于RNA 5’末端非编码区(非翻译区);3)尾部区,位于RNA 3’编码区下游,相当于末端非编码区(非翻译区);4)调控区,包括启动子和增强子等。基因编码区的两侧也称为侧翼顺序。一个典型的模式图:
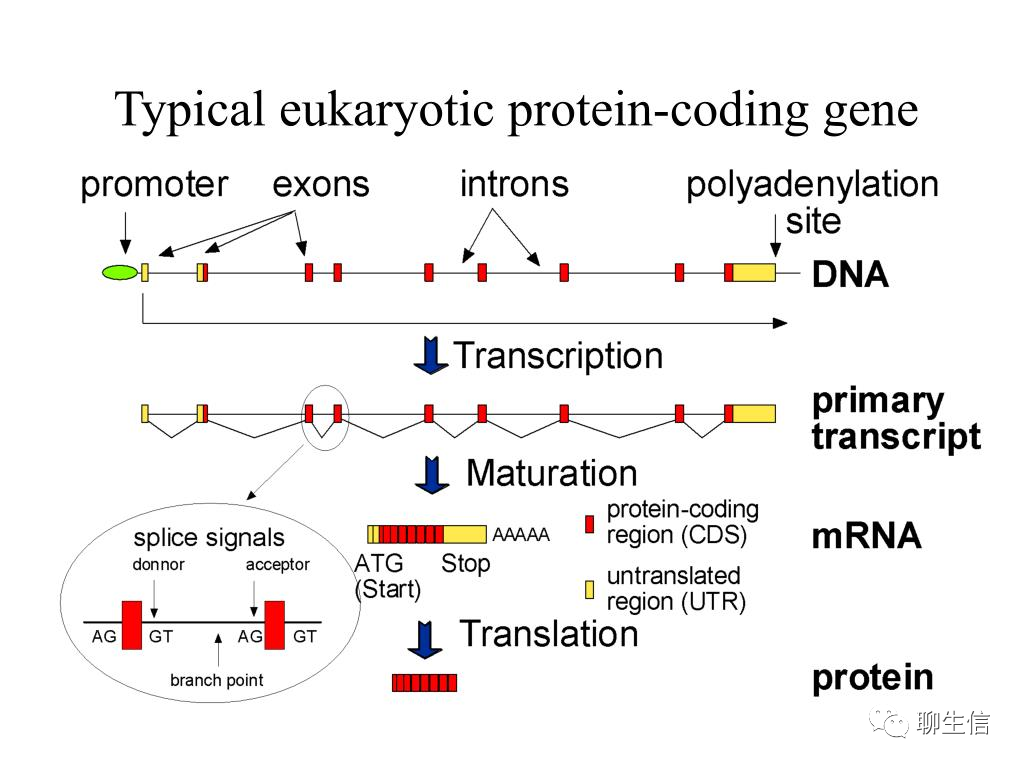
外显子组(Exome):全部外显子称为“外显子组”(Exome)。外显子(Exon)作为真核生物基因的一部分,包含着合成蛋白质(生命活动的承担者)所需要的核心信息。外显子组约占全基因组序列的1%,大多数与疾病相关的变异位于外显子区。与全基因组测序相比,外显子组测序不仅费用较低,数据分析也更为简单,广泛应用于孟德尔遗传病、罕见综合征及复杂疾病的研究中。
全外显子组测序 (Whole Exome Sequencing, WES):是指利用序列捕获技术(主要是核酸探针)将全基因组外显子区域DNA捕捉并富集后进行高通量测序的基因组分析方法。产品主要由Agilent等几家公司把控,不同公司的靶位点略有不同。
变异(Variation):通常指在不同个体、或同一个体的不同细胞之间,基因组或外显子组上的碱基序列的不同。
研究变异的意义。变异位点作为分子遗传标记,在人类复杂疾病、动植物经济性状和育种研究及物种起源、驯化、群体历史动态等方面具有重大的指导意义。所谓指导意义,通常是“一项研究或机制研究的起点”。研究“变异”的一个哲学观或方法论,请查看聊生信之前的一篇评述类文章(点我)。
单核苷酸多态性(Single Nucleotide Polymorphism, SNP):主要是指在基因组水平上由单个核苷酸的变异所引起的DNA序列多态性。多态性是群体基因组或比较基因组研究中的一个概念,有一定的发生和分布规律。因此SNP本身是针对“群体”而言的(within a population),应该在群体中占一定的比例(e.g. > 1%),即一般用来描述一个群体内不那么罕见的碱基突变。
“二态性”(biallelic)。即C>T,C>G,C>A等两种“状态”之间的变异。偶尔也存在三态或四态之间的变异(需要排除测序带来的假阳性)。
SNV(Single Nucleotide Variant):即单核苷酸变异,但频率没有任何限制,可用于描述任意一个可以被测序检测到的碱基突变。除了整个个体或生殖细胞,SNV也可能出现在体细胞中。体细胞的单核苷酸变异(例如肿瘤组织)也可以称为“Single Nucleotide Alteration”。对于少数变异位点的讨论,可直接使用“点突变”(Point Mutation)。
SNP与SNV。二者概念的界限并不是非常明晰,日常交流时甚至一些高水平期刊上也会将这二者混用。SNP更偏向于群体研究,频率可能也较高(但又不绝对,一些SNP的频率也可能极低)。通常SNP数据库的位点总数远低于SNV。在存储SNV的数据时,应尽可能地保留所有样本的全部变异信息(如gVCF文件),这对数据的存储带来了极大的挑战,但对于寻找有意义的罕见位点的变异非常重要。
利用转录组数据分析变异。事实上除了外显子组或基因组,转录组也可以鉴定SNP或SNV等变异。这就要求在设计转录组的实验方案时,小心地将“性状”、“个体”、“器官、组织或细胞”等因素也纳入,最终不仅可以获得基因表达数据,也能获得遗传变异相关的分析结果,并可继续做一些关联分析(如eQTL)。最终测序数据能反映更多的组学信息,提高研究质量。
胚系突变(Germline Variant):又叫生殖细胞突变,是来源于精子或卵子的突变(父母的其它细胞也含有这样的突变),因此通常个体的所有细胞都带有突变。胚系突变可以遗传,一般用于分析遗传病。
新发突变(De novo Mutations):这种突变是指父母本身没有的突变,大多是父母配子(精子、卵子)生成时产生并携带的变异,或受精卵发育过程中的自发突变。每个人身上都会有这样的变异,一般不会带来功能性的问题,但有一些先天的小儿疾病,部分新发突变刚好落在了一个重要的基因上。
体细胞突变(Somatic mutation):又叫获得性突变,是体细胞(如肺、皮肤,肝脏,骨髓,眼睛等)在生长发育过程中或者环境因素影响下后天获得的突变,通常身上只有部分细胞带有突变。体细胞突变通常不会遗传给后代,通常涉及在肿瘤研究中。单有肿瘤样本时无法(直接)区分胚系突变和体细胞突变,只有加入健康样本(健康组织、血液)才能过滤掉胚系突变。
克隆性造血(clonal hematopoiesis):是近几年提出的概念,指造血干细胞亚克隆所携带的突变,可能会对血液样本的WGS或WES变异检测带来一定的影响。克隆性造血的负荷与年龄的增长呈正相关,但突变丰度较低(90%的克隆性造血丰度<1%),跟来源于生殖细胞的胚系突变具有显著差异(胚系突变突变丰度一般在50%或者100%)。但对于肿瘤等体细胞突变研究影响较大,且具有个体特异性,因此必须通过同深度配对的白细胞进行过滤以及优化的生信算法等将其过滤,消除干扰。克隆性造血带有的基因变异一般是非恶性的,且对靶向治疗基因的影响很小,主要发生在DNMT3A、TET2等与靶向治疗无关的基因。
插入缺失(Insertion/Deletion, InDel):是指基因组中小片段(核酸序列)的插入或缺失。
结构变异(Structure Variation, SV):通常指基因组上大长度的序列变化和位置关系变化。基因组结构性变异类型很多,包括长度在50bp以上的长片段序列插入或删除、串联重复、染色体倒位(Inversion)、染色体内部或染色体之间的序列易位(Translocation)、拷贝数变异(CNV)以及复杂的嵌合性变异等。SV也可以发生在两条染色体之间,可使用Circos等软件展示。
拷贝数变异(Copy Number Variation, CNV):是由基因组发生重排而导致的一种染色体结构变异,一般指长度为1 kb以上的基因组大片段的拷贝数增加或者减少(Can be as large as megabases or smaller than 1,000 base pairs), 主要表现为一个群体中的不同个体或同一个体的不同细胞之间亚显微水平的缺失和重复,即数量上与参考基因组或对照组的拷贝数不同。CNV的概念提出只是高通量短序列测序(二代测序)数据分析时,发现有些区间的覆盖度显著高于其它区间(或者是显著低于期望),但通常并不清楚他们被拷贝到了哪些地方,以及是作为整体还是被分段拷贝。
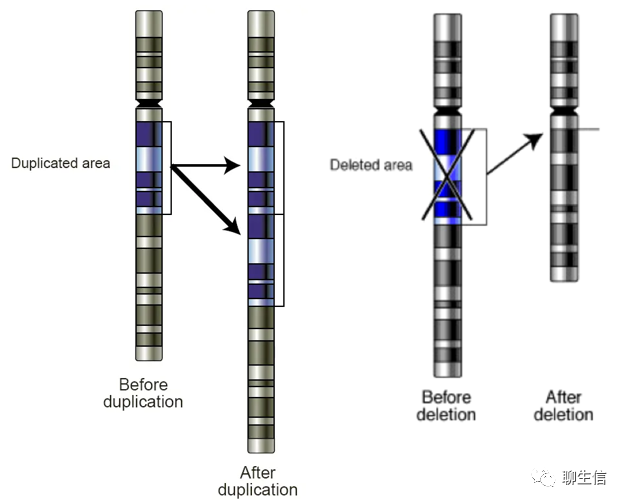
CNV的分类与分布:二倍体生物的CNV可分为:正常(2拷贝)、缺失(1或0拷贝)和重复(>2拷贝)。CNV在染色体上的存在形式主要有:2条同源染色体拷贝数同时出现缺失(或同时出现重复);1条同源染色体发生缺失,另1条正常(或重复);1条同源染色体出现拷贝数重复,另1条正常。
InDel vs CNV。目前主流的相关分析工具(BWA,bowtie2等)和算法(Smith-Waterman的local-alignment等)能够直接鉴定出来的插入和删除(InDel),检测的范围一般是从1bp到50bp。至于更大尺度的丢失和获得,主要是通过分析序列的覆盖度鉴定为CNV。
拷贝数多态性(Copy Number Polymorphism, CNP):一个CNV在群体中的频率超过1%时通常成为CNP。类似的比较可参考上文的SNP vs SNV。
SD区域(Segment Duplication Region)或串联重复区域(Tandem Repeat Region):串联重复是由序列相近的一些DNA片段串联组成。例如在人类染色体22号和Y染色体上的大量SD序列。
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|



























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集