
Science封面| 谷歌实现全球首个量子化学模拟,用量子「计算」出化学反应过程
新智元
共 2838字,需浏览 6分钟
·
2020-08-30 13:51

新智元报道
新智元报道
来源:science
编辑:白峰
【新智元导读】化学键是如何形成的?由于庞大的计算量和分子的复杂性,经典的计算方法难以揭示化学反应的真实过程。但最近,谷歌AI的研究团队成功把量子计算应用在了化学反应的模拟上,荣登Science封面。

Hartree-Fock:实现量子化学计算的核心「构建块」
Hartree-Fock模型:凝聚态物理中的「电子-电子相互作用」是多体问题,无法获得解析解。因此需要一种能近似计算电子-电子相互作用能的方法,Hartree-Fock 方法就是其中的一种。其核心思路是平均场近似,将一个电子受其他所有电子的作用和用一个等效的场来表示。
遍历数千节点, Sycamore实现高保真量子计算
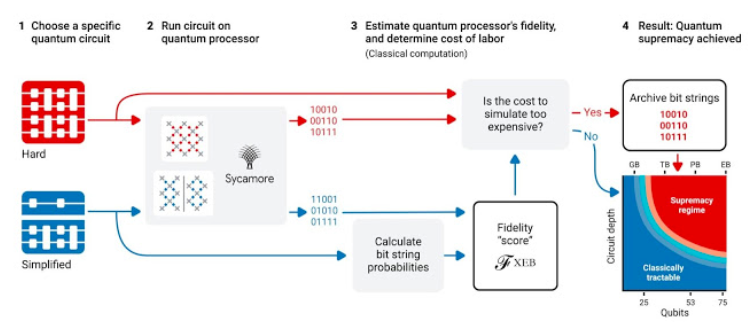 利用Sycamore展示量子计算优势
利用Sycamore展示量子计算优势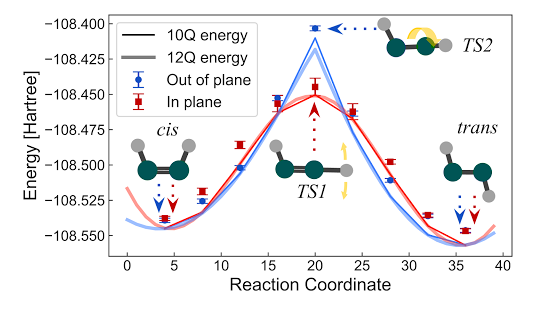
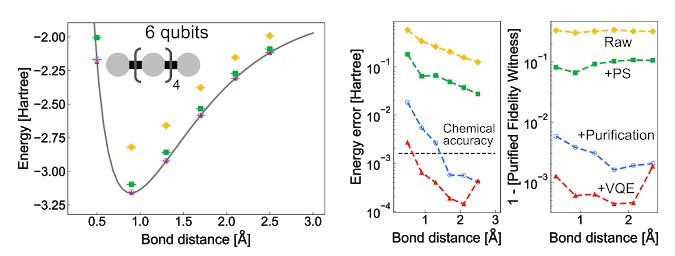

量子化学:量子力学和化学的交叉产物


评论