哈佛大学单细胞课程|笔记汇总 (三)
生物信息学习的正确姿势
NGS系列文章包括NGS基础、在线绘图、转录组分析 (Nature重磅综述|关于RNA-seq你想知道的全在这)、ChIP-seq分析 (ChIP-seq基本分析流程)、单细胞测序分析 (重磅综述:三万字长文读懂单细胞RNA测序分析的最佳实践教程)、DNA甲基化分析、重测序分析、GEO数据挖掘(典型医学设计实验GEO数据分析 (step-by-step))、批次效应处理等内容。
(三)Single-cell RNA-seq: Quality control set-up
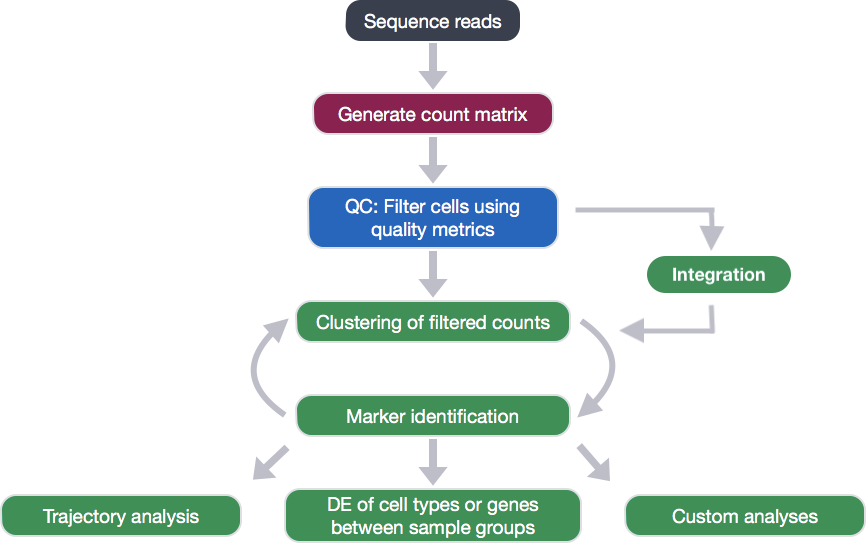
在生成count矩阵后,我们需要对其进行QC分析,并将其导入R进行后续分析:
探索示例集
该数据集来自Kang et al, 2017(https://www.nature.com/articles/nbt.4042)的文章,是八名狼疮患者的PBMC(`Peripheral Blood Mononuclear Cells`)数据,将其分为对照组和干扰素beta处理(刺激)组。
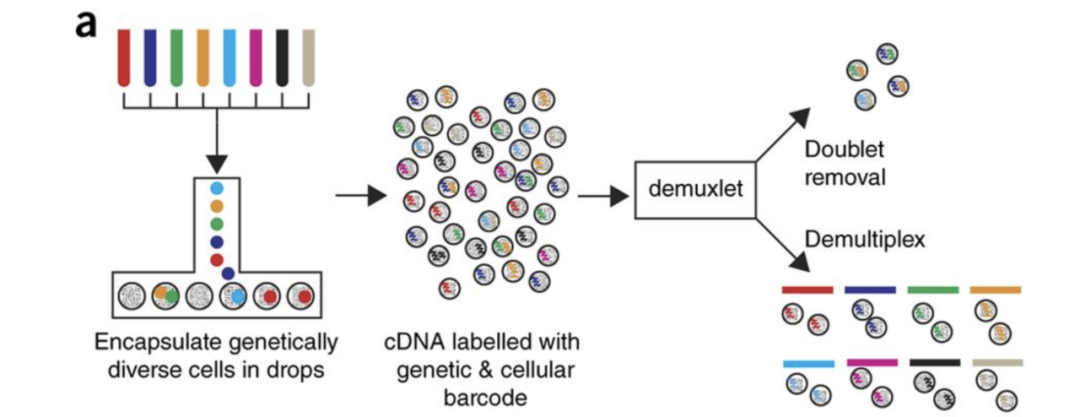
Image credit: Kang et al, 2017(https://www.nature.com/articles/nbt.4042)
Raw data
研究团队发现GEO (GSE96583:https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE96583)提供的矩阵缺少线粒体的reads,因此从SRA (SRP102802:https://www-ncbi-nlm-nih-gov.ezp-prod1.hul.harvard.edu/sra?term=SRP102802)下载BAM文件,然后转化为FASTQ文件,并使用`Cell Ranger`(https://support.10xgenomics.com/single-cell-gene-expression/software/pipelines/latest/what-is-cell-ranger)获得count矩阵。
NOTE: The counts for this dataset is also freely available from 10X Genomics and is used as part of the
Seurat tutorial(https://satijalab.org/seurat/v3.0/immune_alignment.html).(我说我咋这么眼熟。。。)
Metadata
样品属性相关信息也叫metadata。关于以上数据的metadata如下:
使用10X Genomics版本2化学试剂制备文库;
样品使用Illumina NextSeq 500测序;
将来自八名狼疮患者的PBMC样本分别分成两等份:
一份用100 U/mL重组IFN-β激活PBMC,处理时间为6小时。
另一份样品未经处理。
6小时后,将不同条件的8个样品一起收集起来(受激细胞和对照细胞)。
分别鉴定了
12138和12167个细胞(去除doublets后)作为对照样品和刺激后的合并样品。由于样品是PBMC,因此我们期望有免疫细胞,例如:
B cells
T cells
NK cells
monocytes
macrophages
possibly megakaryocytes
It is recommended that you have some expectation regarding the cell types you expect to see in a dataset prior to performing the QC. This will inform you if you have any cell types with low complexity (lots of transcripts from a few genes) or cells with higher levels of mitochondrial expression. This will enable us to account for these biological factors during the analysis workflow.
上述细胞类型都不预估有低复杂度 (少部分基因表达大部分转录本)或高线粒体含量。
设置R环境
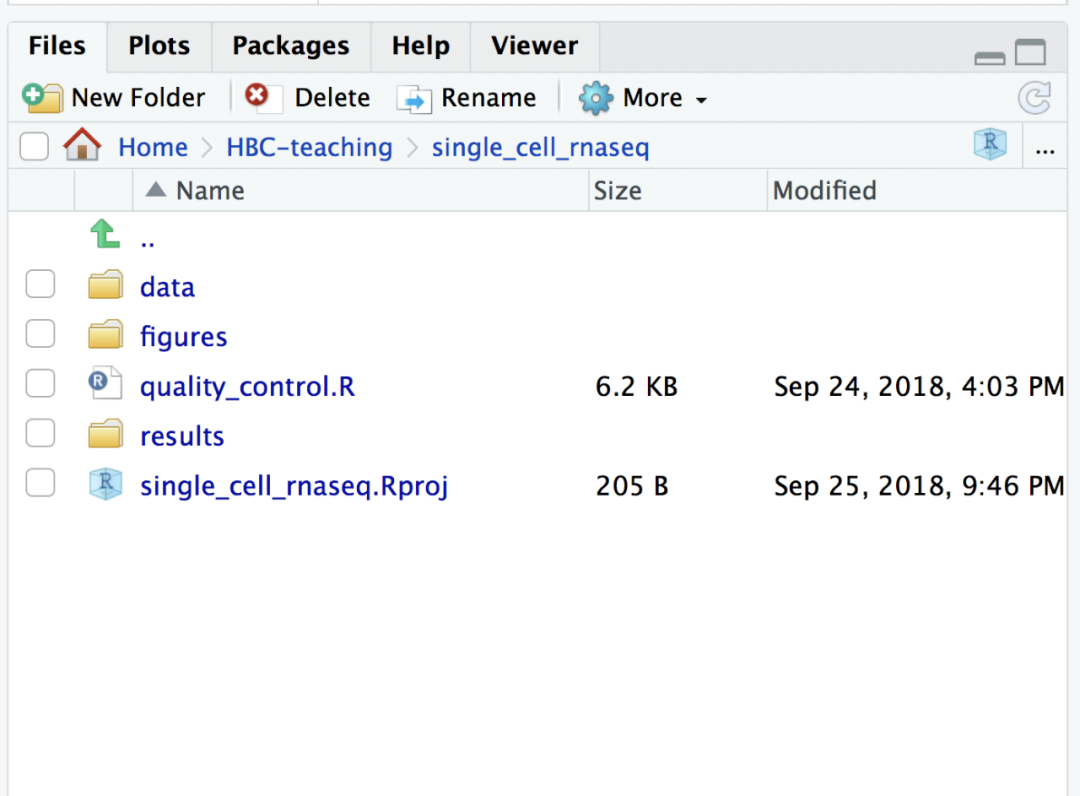
为了更好的管理数据,使得整个项目具有组织性,先创建一个项目叫“single_cell_rnaseq”,然后构建以下目录:
single_cell_rnaseq/
├── data
├── results
└── figures
下载数据
Control sample (https://www.dropbox.com/sh/73drh0ipmzfcrb3/AADMlKXCr5QGoaQN13-GeKSa?dl=1)
Stimulated sample (https://www.dropbox.com/sh/cii4j356moc08w5/AAC2c3jfvh2hHWPmEaVsZKRva?dl=1)
解压数据后新建一个R脚本,命名为“quality_control.R”,并且保证整个的工作目录看起来像这样:
加载R包
library(SingleCellExperiment)
library(Seurat)
library(tidyverse)
library(Matrix)
library(scales)
library(cowplot)
library(RCurl)
加载count矩阵
一共有3个文件,分别为:cell IDs、gene IDs和matrix of counts(每个细胞的每个基因)。
barcodes.tsv:这是一个文本文件,其中包含该样品存在的所有细胞条形码。条形码以矩阵文件中显示的数据顺序列出(这些是列名)。

features.tsv:这是一个文本文件,其中包含定量基因的标识符。标识符的来源可能会有所不同,具体取决于定量过程中使用的参考(即Ensembl,NCBI,UCSC),大多数情况下这些都是官方gene symbol。这些基因的顺序与矩阵文件中各行的顺序相对应(这些是行名)。

matrix.mtx:注意该矩阵中有许多zero values。
有两种方法可以进行矩阵读入,分别为readMM()和Read10X()。
readMM(): This function is from the Matrix package and will turn our standard matrix into a sparse matrix. The features.tsv file and barcodes.tsv must first be individually loaded into R and then they are combined. For specific code and instructions on how to do this please see our additional material(https://github.com/hbctraining/scRNA-seq/blob/master/lessons/readMM_loadData.md).
Read10X(): This function is from the Seurat package and will use the Cell Ranger output directory as input. In this way individual files do not need to be loaded in, instead the function will load and combine them into a sparse matrix for you. We will be using this function to load in our data!
Reading in a single sample (read10X())
我们通常使用Read10X()。原因就要从头说起了,一般在软件Cell Ranger处理10X数据后会生成一个outs目录,在outs文件夹中有这样几个文件:
web_summary.html: 包括许多QC指标,预估细胞数,比对率等;
BAM alignment files:用于可视化比对的reads和重新创建
FASTQ文件;filtered_feature_bc_matrix: 经过
Cell Ranger过滤后构建矩阵所需要的所有文件;raw_feature_bc_matrix: 未过滤的可以用于构建矩阵的文件;
如果你有一个单个样本的话,可以直接构建a Seurat object(https://github.com/satijalab/seurat/wiki/Seurat):
# How to read in 10X data for a single sample (output is a sparse matrix)
ctrl_counts <- Read10X(data.dir = "data/ctrl_raw_feature_bc_matrix")
# Turn count matrix into a Seurat object (output is a Seurat object)
ctrl <- CreateSeuratObject(counts = ctrl_counts,
min.features = 100)
NOTE: The min.features argument specifies the minimum number of genes that need to be detected per cell. This argument will filter out poor quality cells that likely just have random barcodes encapsulated without any cell present. Usually, cells with less than 100 genes detected are not considered for analysis.
在使用Read10X()函数读取数据时,Seurat会为每个细胞自动创建metadata,存储在meta.data slot。
The Seurat object is a custom list-like object that has well-defined spaces to store specific information/data. You can find more information about the slots in the Seurat object at this link:https://github.com/satijalab/seurat/wiki/Seurat.
# Explore the metadata
head(ctrl@meta.data)
metadata 的每一列代表什么呢?
orig.ident: 细胞聚类的cluster,含有样本的身份信息(已知的情况下);nCount_RNA: 每个细胞的UMI数目;nFeature_RNA: 检测到的每个细胞中的genes数目。
使用for循环对多个数据进行读入
# Create each individual Seurat object for every sample
for (file in c("ctrl_raw_feature_bc_matrix", "stim_raw_feature_bc_matrix")){
seurat_data <- Read10X(data.dir = paste0("data/", file))
seurat_obj <- CreateSeuratObject(counts = seurat_data,
min.features = 100,
project = file)
assign(file, seurat_obj)
}
于是分别形成了ctrl_raw_feature_bc_matrix和stim_raw_feature_bc_matrix两个seurat对象,使用merge函数对不同数据进行合并:
# Create a merged Seurat object
merged_seurat <- merge(x = ctrl_raw_feature_bc_matrix,
y = stim_raw_feature_bc_matrix,
add.cell.id = c("ctrl", "stim"))
因为相同的细胞ID可以用于不同的样本,所以我们使用add.cell.id参数为每个细胞ID添加特定于样本的前缀。如果我们查看合并对象的metadata,我们应该能够在行名中看到前缀:
# Check that the merged object has the appropriate sample-specific prefixes
head(merged_seurat@meta.data)
tail(merged_seurat@meta.data)
往期精品(点击图片直达文字对应教程)
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|
|



























后台回复“生信宝典福利第一波”或点击阅读原文获取教程合集